Синдром ангельмана фото, Синдром Ангельмана: характерные клинические признаки — Московское общество детских врачей

Диагноз был поставлен исключительно на основе клинических проявлений, ведь с помощью доступных лабораторных исследований, врач не смог тогда предоставить научных доказательств того, что эти трое детей были больны одной болезнью. Это может быть связано с генетическими ошибками, такими как делеции или мутации в сегменте ой хромосомы, однородительская дисомия или транслокации. Я нахожу это абсолютно оскорбительным и не понимаю, как кто-то в сообществе слепых может принять ее выбор Кармен 45 лет, мама с двумя детьми. В исследование были включены 60 детей 37 девочек и 23 мальчика с синдромом Ангельмана, которых наблюдали на протяжении от 6 мес до 2-х лет.
Синдром Ангельмана в большинстве случаев развивается, когда по каким-то причинам мамин UBE3A малышу не достаётся. Или достаётся, но не работает. Впрочем, это не единственная причина. Как правило, в таком случае дети имеют нарушения, связанные с другими генами или хромосомами. Этот тест позволяет оценить состояние хромосом, обнаружить в них повреждения или аномалии, а также «вычислить» работоспособность UBE3A и определить, от кого он достался — от матери или от отца.
Как и большинство генетических заболеваний, синдром Ангельмана неизлечим. Учёные ведут исследования в надежде научиться чинить или восстанавливать гены. И возможно, однажды появятся лекарства для подобных генетических нарушений. На сегодня же лечение синдрома Ангельмана сводится к тому, чтобы облегчить ребёнку жизнь и по возможности помочь ему адаптироваться в окружающем мире.
С таким малышом обычно работают сразу несколько специалистов, а реабилитация включает в себя:. Если с малышом заниматься, он станет менее возбудимым, будет лучше спать, сможет играть с другими детьми. Вряд ли у него получится учиться в обычной школе. Однако общаться с окружающими и ухаживать за собой — например, выполнять простые гигиенические процедуры, есть с помощью ножа и вилки, одеваться и раздеваться, заниматься несложной деятельностью взрослому человеку с синдромом Ангельмана вполне под силу.
В силу особенностей нервной системы ребёнок с синдромом не способен оценить своё состояние. И постоянно нуждается в поддержке. Поэтому основная нагрузка — и физическая, и прежде всего психологическая — ложится на его родителей. Это тяжело. Поэтому врачи настоятельно рекомендуют мамам и папам искать поддержку. Надо брать: ультратонкий пауэрбанк Baseus Blade 2 за 4 рублей. Находки AliExpress: подставка для ноутбука, краскопульт и спидометр для велосипеда.
Лайфхак: как купить ноутбук, чтобы не нарваться на брак или подделку. По стопам Кэрри Брэдшоу и Паддингтона: 6 мини-экскурсий по культовым книгам и фильмам. Спорт и фитнес.
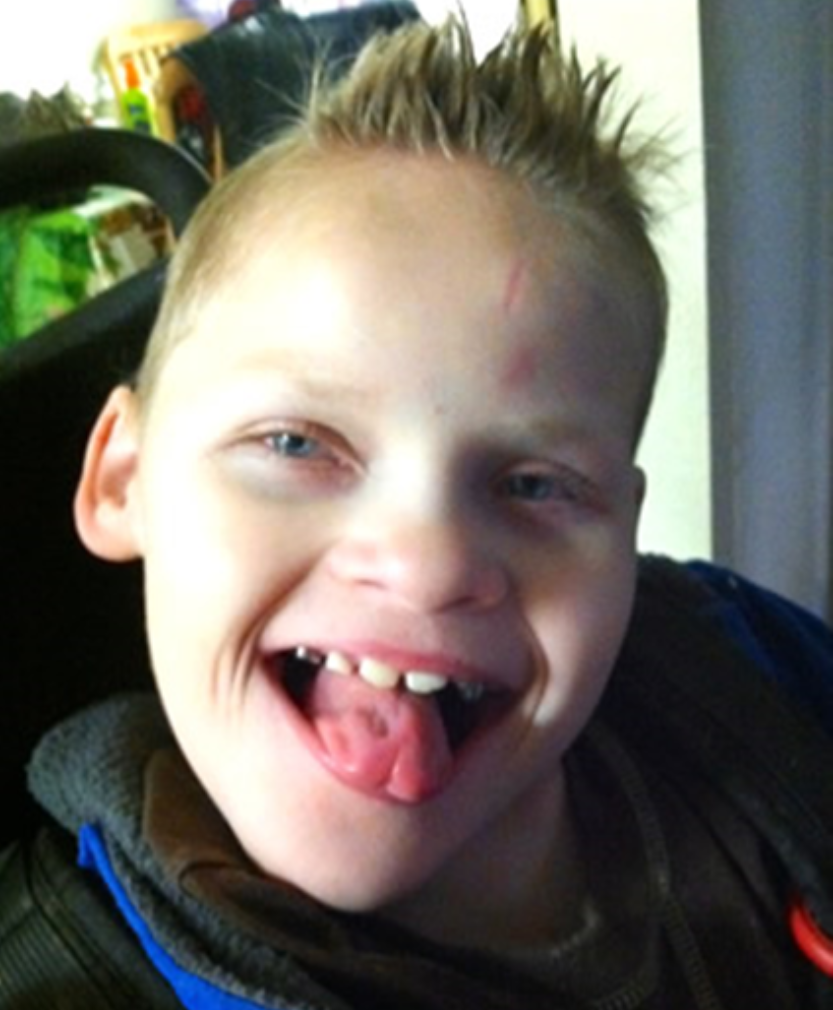
Ликбез Здоровье 24 февраля Тот случай, когда счастливый детский смех является нехорошим симптомом. Екатерина Комиссарова Медицинский журналист Лайфхакера. Что такое синдром Ангельмана Синдром Ангельмана — это редкое генетическое нарушение, при котором страдает нервная система.
Лучшие предложения. Это интересно.

Лайфхак: как купить ноутбук, чтобы не нарваться на брак или подделку Реклама. Как мужчине подготовиться к отцовству и зачем это нужно делать Социальная реклама. Станьте первым, кто оставит комментарий. У здорового человека, экспрессия материнской аллели - сильнее, тогда как отцовская аллель почти не проявляется. Если же материнские копии генов - удалены или мутированы, то это вызывает появление синдрома Ангельмана если вследствие аналогичных механизмов потеряна отцовская копия , то это вызывает синдром Прадера-Вилли.
Следует отметить, что тест на метилирование, который проводится для диагностики синдрома Ангельмана дефект UBE3A на самом деле направлен на поиск соседнего гена SNRPN который имеет противоположный характер метилирования.
Синдром Ангельмана может также быть результатом мутации одного гена. Этот ген UBE3A , участвующего в метаболизме убиквитина присутствует на обоих копиях хромосом на родительской и на материнской , но его действие отличается от процесса метилирования импринтинга. Инактивация отцовской копии гена UBE3A происходит в мозге в гиппокампе и мозжечке , тогда как материнская аллель почти всегда остается активной.
Наиболее распространенным генетическим дефектом , который приводит к появлению синдрома Ангельмана, является: делеция 4Mb мега материнской копии хромосомной области 15q , которая вызывает отсутствие экспрессии родительской копии UBE3A в мозге.
И именно выявление четырех субстратов Е6-AP позволило несколько понять возможные молекулярные механизмы, лежащие в основе возникновения синдрома Ангельмана.
Начальные исследования, которые проводятся с помощью мышей, у которых отсутствует экспрессия материнской копии гена UBE3A, обнаружили серьезные отклонения в деятельности гиппокампа , которые нарушают процесс формирования памяти. В частности, наблюдаются отклонения в парадигмах обучения, особенно в тех процессах, которые зависят от деятельности гиппокампа - кондиционирование страхом.
Это позволяет обеспечить связь между искусственной синаптической пластичностью гиппокампа и формированием гиппокамп-зависимой памяти в естественных условиях, и молекулярной природе синдрома Ангельмана. Клинические признаки Ниже описаны характерные для синдрома Ангельмана клинические характеристики и частота их распространенности среди больных: 1.
Одной из наиболее примечательных особенностей синдрома Ангельмана является его патогномонические нейрофизиологические характеристики. Зачастую на электроэнцефалограмме появляются очень велоамплитудные ритмы с частотой 2 -3 Гц, крупнейшими являются отклонения в префронтальной зоне.
Следующим наиболее распространенным нарушением является симметрический високовольтажный ритм частотой Гц. Третья характерная картина, Гц активность чередуется с острыми пиковыми волнами в затылочных отделах, и связан с закрытием глаз. Приступы смеха, никак не влияют на ЭЭГ и рассматриваются, исключительно как такие, которые не имеют никакого отношения к ЭЭГ. Лечение и уход. На сегодня отсутствуют доступные эффективные лекарства для преодоления синдрома Ангельмана.
То есть все методы лечения направлены на поддержание нормального состояния больного и является, преимущественно, симптоматическими. Так, эпилепсию можно контролировать с помощью одного или нескольких типов противосудорожных лекарств. Однако, при определении типов противосудорожных препаратов, которые необходимы для симптома, возникают определенные трудности, которые обычно связаны с наличием нескольких типов приступов судорог, а не одного, как при обычной эпилепсии.
Многие больные употребляют мелатонин , для лучшего сна, ведь расстройство часто влияет именно на процесс сна. Многие люди с синдромом Ангельмана спят не более 5 часов в день.
Для поддержки, стимуляции нормальной работы кишечника часто используются мягкие слабительные средства, а осуществление физиотерапии на ранней стадии позволяет поддержать функциональность суставов.
Больные СА, как правило, люди, которые имеют счастливый вид, они любят общаться с людьми и играть в разные игры. Однако, установление связи с другими людьми, общение с ними может сначала быть несколько трудным, но, поскольку дети с СА постепенно развиваются, у них возникает определенный характер, появляются разные способности и их начинают лучше понимать другие люди. В частности, люди с синдромом Ангельмана очень часто применяют невербальные средства общения , для того, что бы компенсировать ограниченные возможности речи.
Часто, врачи отмечают, что их понимание развито гораздо больше, чем коммуникационные способности. Большинство больных, применяют, как правило, лишь слов, если вообще разговаривает. Приступы судорог и беспричинный смех также являются последствиями развития заболевания. Тяжесть симптомов, возникающих при синдроме Ангельмана, отличаются в каждом конкретном случае. Некоторые люди, с легкой формой синдрома Ангельмана , имеют большой словарный запас и высокую степень самоконтроля.
Однако ходьба и использования простых языковых знаков может пострадать значительно сильнее. Кроме того, считается, что определенный генетический механизм, лежащий в основе расстройства, коррелирует с общим прогнозом для потерпевшего. Так, мутации в гене UBE3A , связанны с наименьшим влиянием болезни на организм , а крупные делеции 15 хромосомы - хуже влияют на больных СА.
Клинические признаки синдрома Ангельмана с возрастом меняются. В частности, реже встречаются такие проявления, как гиперактивность и плохой сон. Приступы судорог, тоже встречаются реже или вообще прекращаются, равно как и аномалии ЭЭГ. Для снижения частоты возникновения судорог, врачи рекомендуют употреблять специальные лекарственные препараты.
Также следует отметить то, что частота и тяжесть приступов судорог часто влияет на процесс полового созревания девочек, больных СА, это влияние только краткосрочное.
Черты лица меняются не существенно, однако многие взрослые с СА выглядят очень молодыми для своего возраста. Половое созревание и менструация начинается в нормальном возрасте. Половое развитие, как считают врачи, существенно не нарушается, о чем свидетельствует известный случай женщины с синдромом Ангельмана, которая родила ребенка женского пола, также больного этим синдромом. У большинства пациентов с СА наблюдается недержание мочи.
Синдром Ангельмана - это не нейродегенеративный синдром, то есть его проявления можно уменьшить, если проводить необходимое лечение. Для больных синдромом Ангельмана следует выбирать одежду без молний и кнопок, ведь только в таком случае они смогут одеться сами.
Большинство взрослых могут есть ножом или ложкой и вилкой, а также могут научиться выполнять простую домашнюю работу. Общее состояние здоровья достаточно хорошее, продолжительность жизни - средняя. Характерные проблемы , возникающие у взрослых больных СА это: ожирение больше у женщин , ускоренное прогрессирование сколиоза если он присутствует.
Ласковый характер, который является очень положительной чертой для маленьких детей, может иметь не очень хорошие последствия во взрослой жизни, однако эту проблему можно решить, окружив больных доброжелательной атмосферой и обеспечив соответствующее лечение и уход.
Дальтонизм часть вторая Дальтонизм часть первая Дефицит 3 гидрокси-метил-глутарил-КоА лиазы Дефицит 3-метилкротонил-коэнзим А карбоксилазы Дефицит альфа 1 антитриптисина Дефицит бета-кетотиолазы Дефицит биотинидазы Дефицит дигидропиримидин дегидрогеназы Дефицит длинноцепочечной ацил-коэнзим А дегидрогеназы Дефицит короткоцепочечной ацил-коэнзим А дегидрогеназы Дефицит метилентетрагидрофолат редуктазы Дефицит прекалликреин Дефицит синтетазы голокарбоксилазы Дефицит фактора ХI Иминоглицинурия Муковисцидоз Мышечная дистрофия Дюшенна Нейрофиброматоз Органические ацидемии Первичный системный дефицит карнитина Перманентный неонатальный сахарный диабет Перонеальная дистрофия Шарко-Мари-Тута часть вторая Перонеальная дистрофия Шарко-Мари-Тута часть первая Пропионовая ацидурия Псевдополидистрофия Гурлера Семейная вегетативная дисфункция Семейная гиперхолестеринемия.

Цитрулинемия Энтеропатический акродерматит. Основы генетики.